Introduction
Alzheimer’s disease (AD) should need no introduction. It is a neurodegenerative disorder with a prevalence of 10-30% in the population over 65 years of age (Kawas et al., 2000), causing devastating effects both subjectively, on patients’ and caregivers’ lives, and objectively, on national healthcare costs.
First observed and reported by Dr Alois Alzheimer in 1906 (Sahyouni, Verma & Chen, 2016, p. 121), the hallmark features in the pathophysiology of the disease are Amyloid plaques and neurofibrillary tangles (NFTs). The former are accumulations of insoluble forms of the amyloid-β (Aβ) protein in the extracellular space, while the latter are aggregations of anomalous hyperphosphorylated forms of the microtubule protein tau in intracellular spaces.
Both these features have historically been associated with the widespread neurodegeneration seen in the disease, progressing through neuronal cell death and eventually leading to cortical atrophy (Figure 1).
](figure_1.png)
The disease usually first spreads from the hippocampus and the temporal lobes to areas of the neocortex. Notably, the clinical phase of the disease is preceded by a largely asymptomatic phase that is thought to last as long as two decades (Villemagne et al., 2013), during which amyloid starts accumulating but cognitive impairment is mild if at all present. The clinical duration of Alzheimer’s is of 8-10 years, with patients usually dying for complications associated with the disease (in two-thirds of patients this is pneumonia due to cough reflex suppression. Sahyouni, Verma & Chen, 2016, p. 93-94), after having largely lost cognitive functions, self-sufficiency and memories of loved ones.
As the world population ages, the threat represented by the disease grows in importance. In this essay, I am going to discuss the prominent theories of the causes of Alzheimer’s, to see in what direction this vital research area is headed.
The amyloid cascade hypothesis
The amyloid-β peptide is a metabolic product of the amyloid precursor protein (APP), which is a transmembrane glycoprotein thought to have some roles in synaptic plasticity (Masters et al., 2015). APP can be cleaved following two pathways; when cleaved successively by β- and γ-secretase, Aβ is released. Aβ peptides can be generated at various lengths. The most abundant form, Aβ1-40 is sufficiently soluble to be usually cleared before depositing. On the other hand, Aβ1-42 (and more rarely Aβ1-43) fragments are less soluble and can readily aggregate to build from toxic oligomers (Benilova, Karran & De Strooper, 2012) to, ultimately, plaques.
The idea that accumulation of Aβ is a key event in the pathogenesis of Alzheimer’s is known as the “amyloid cascade hypothesis” (Hardy & Higgins, 1992), and for many years has been the leading theoretical framework used by researchers to understand the disease. Aβ overproduction or lack of clearance is thought to evoke a cascade of responses which ultimately leads to neuronal loss and dementia (Figure 2). Specifically, the cytotoxic effects of Aβ are now thought to be mediated mainly by Aβ1-42 oligomers, which are associated, among other things, with calcium dyshomeostasis, mitochondrial damage (Waldemar & Burns, 2017, p. 11-12), decrease in synaptic density and tau hyperphosphorylation (Selkoe & Hardy, 2016).
Evidence supporting the hypothesis comes mainly from genetic studies. The APP gene is located on chromosome 21. Since it has been observed that individuals with Down syndrome all typically get AD by 35 years of age (Wisniewski, Wisniewski & Wen, 1985), this suggests that it might be due to the Aβ overproduction caused by APP overexpression. This has been corroborated by various further studies (Prasher et al., 1998; Rovelet-Lecrux et al., 2006).
Mutation in the genes coding for presenilin 1 and 2, parts of the γ-secretase complex cleaving APP, have also been strongly linked with AD, specifically with increased production of Aβ1-42 oligomers (Kumar-Singh et al., 2006).
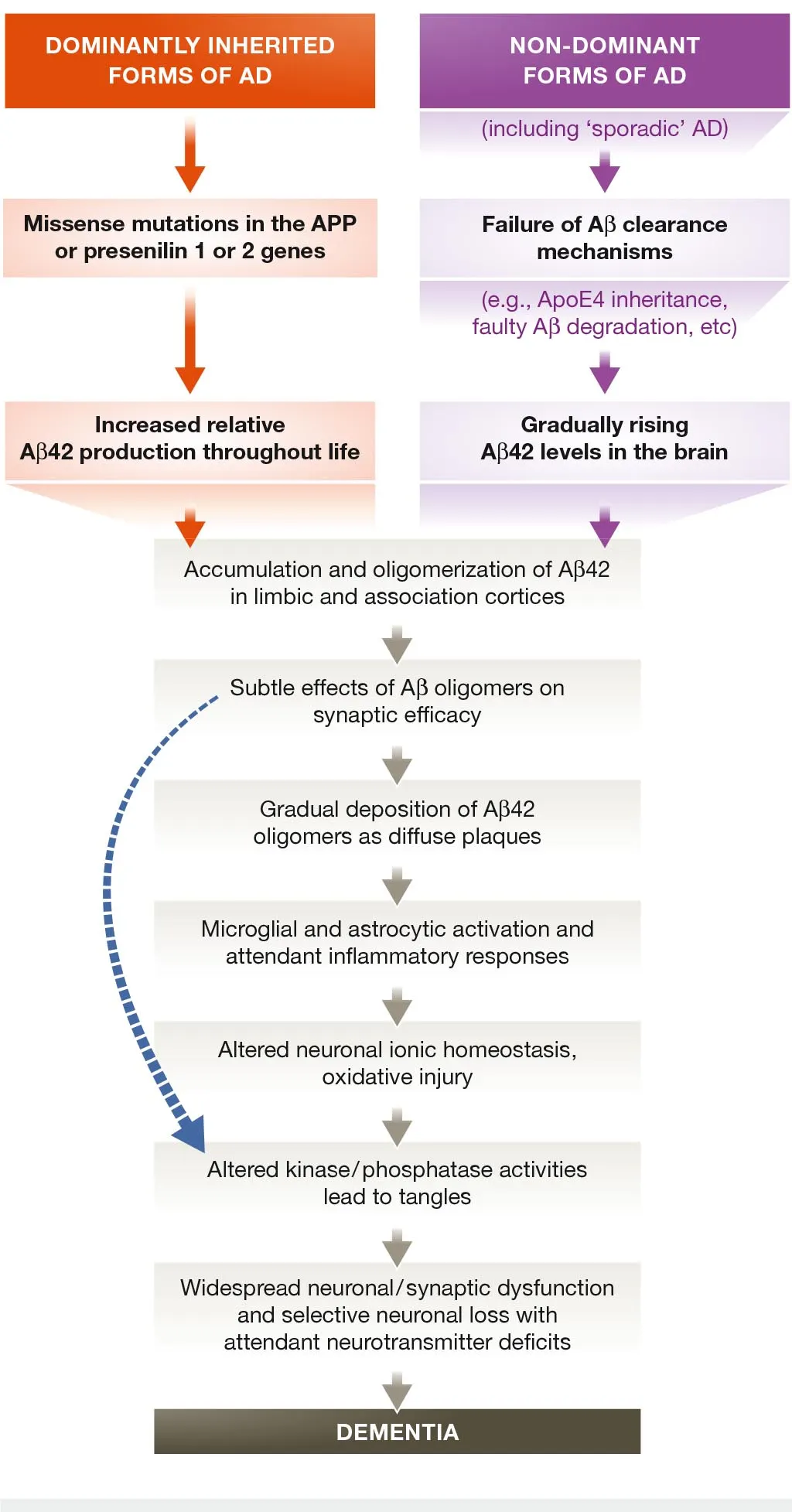
Furthermore, an APP missense mutation has been identified that alters Aβ production and properties (Jonsson et al., 2012). This mutation has been found to be protective against plaque formation, AD development and even age-related decline in cognitive functions.
Challenging the amyloid cascade hypothesis
The merit of the hypothesis has been to generate testable predictions along the years and to focus the efforts of research (Hardy, 2009), but recent evidence now suggests it might be time for a change in paradigm.
To begin with, large clinical trials of many different drugs trying to tackle Aβ production, aggregation or clearance have failed to show benefits in patients with mild to moderate AD (Panza et al., 2019).
The current effort is directed more towards Aβ oligomers, but some observe that brains in which plaques are present must have been exposed to oligomers for a long time (Panza et al., 2019). This is problematic when we consider that amyloid plaques can be observed in cognitively healthy individuals (Herrup, 2015).
What is more, research is starting to appreciate the physiological roles of amyloid-β. It appears in fact that brain amyloid levels are raised in an impressive variety of conditions, from traumatic brain injury (Marklund et al., 2014) to sleep deprivation (Ju et al., 2017). Aβ could serve a broad role in modulating synaptic function (Brothers, Gosztyla & Robinson, 2018), and its secretion might represent the brain’s attempt to mitigate neuronal damage (Panza et al., 2019). A better appreciation of the physiological roles of Aβ might come to explain why γ-secretase inhibitors ended up worsening AD symptoms in some clinical trials (Doody et al., 2013).
However, at this point in the discussion – parallel to what happened for some time in the research world – we have lost focus of another key player: the microtubule protein tau.
The role of tau
A key passage in the amyloid cascade hypothesis is the alleged link between Aβ and tau.
Tau stabilizes microtubules and enables axonal transport in healthy neurons. Cycles of tau phosphorylation and de-phosphorylation allow the movements of cargo containing substances such as nutrients and neurotransmitters along the axon. However, if tau becomes abnormally phosphorylated, it can start to accumulate inside the neuron and form NFTs, with disrupting effects (Waldemar & Burns, 2017, p. 12-13).
Unlike amyloid, tau alone is sufficient to cause neurodegeneration and is involved in a wide spectrum of diseases, from frontotemporal dementia to Pick’s disease, aptly referred to as tauopathies (Kovacs, 2018, Chapter 25). Even in AD, the advance of neurodegeneration correlates much more strongly with tau rather than Aβ pathology (Ossenkoppele et al., 2016).
A question to address remains: is tau degeneration just a more proximal cause, with Aβ upstream in the cascade?
A complex interaction
Determining the nature of the interaction between Aβ and tau is perhaps the biggest challenge that research on AD has faced and will continue to face. Evidence is, to say the least, disorienting.
On the one hand, we have a series of studies suggesting that Aβ is upstream of tau in the disease pathology. Those are summarised in Table 1.
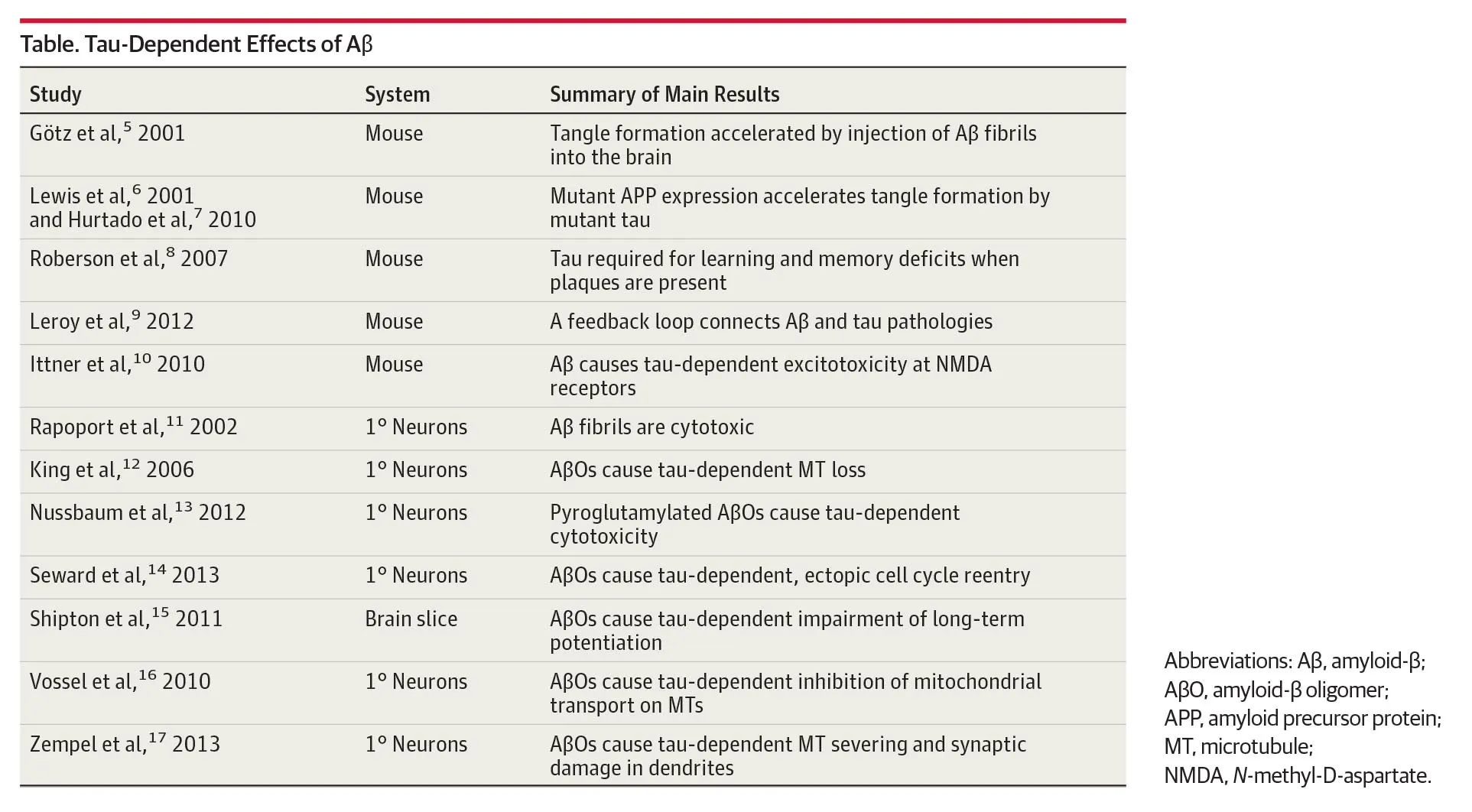
On the other hand, there is the puzzling finding that Aβ and tau accumulation originates in different areas of the brain (van der Kant, Goldstein & Ossenkoppele, 2019). The former in the association cortices and the latter in the trans-entorhinal cortex. This has been termed the “spatial paradox” and is shown in Figure 3.
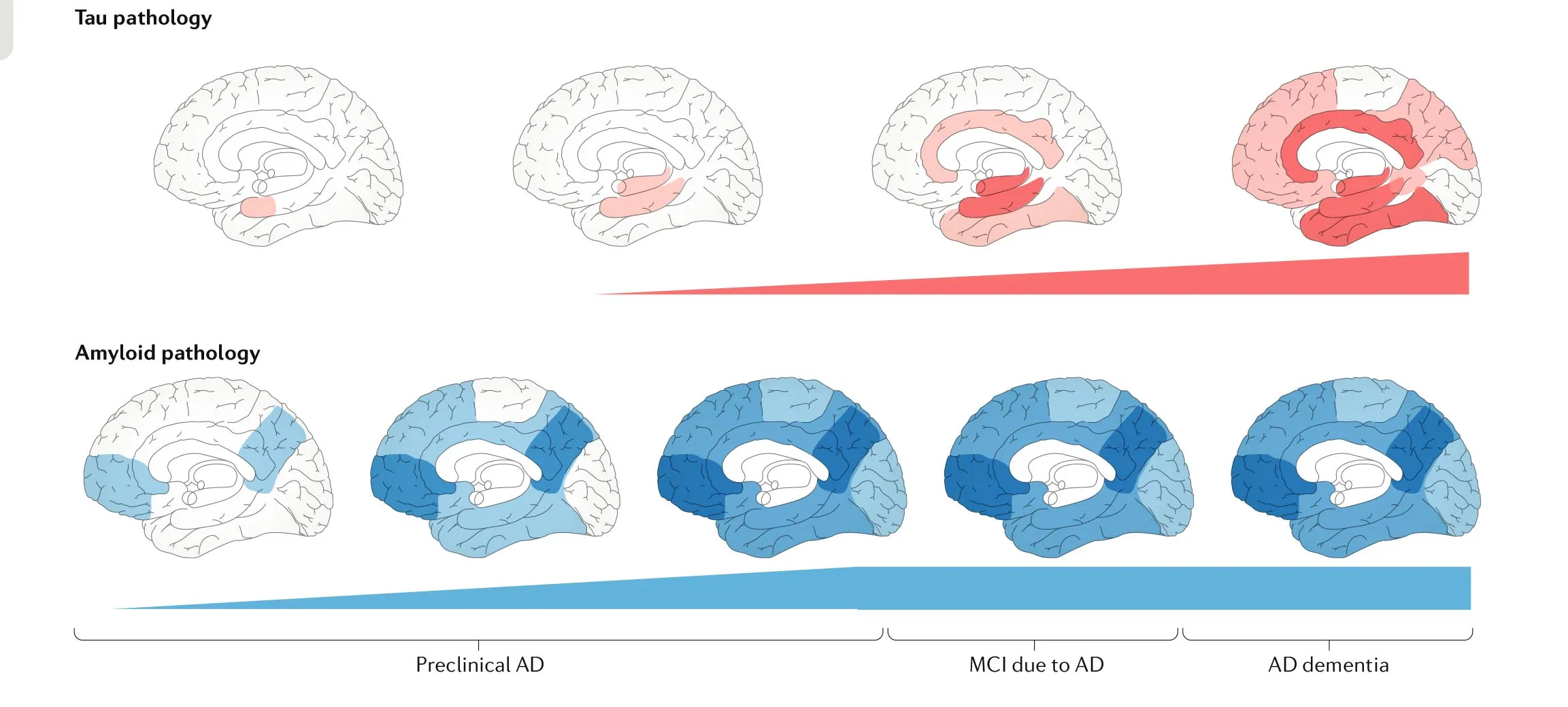
This might be explained by the discovery of some independent regulators of both Aβ and tau pathology: the lipid metabolism, the endocytic system and the brain immune system (van der Kant, Goldstein & Ossenkoppele, 2019).
Interestingly, however, tau does not seem to be able to spread alone to neocortical regions in the absence of Aβ pathology (Jack et al., 2018). On the light of this, researchers are now inclined to conclude that Alzheimer’s disease is an Aβ or APP facilitated tauopathy (Kametani & Hasegawa, 2018; van der Kant, Goldstein & Ossenkoppele, 2019).
However, to put the amyloid cascade hypothesis to a final test, researchers should include tau PET measures in clinical trials of Aβ-targeting drugs, since tau monitorization has not been efficient in the past (Figure 4).
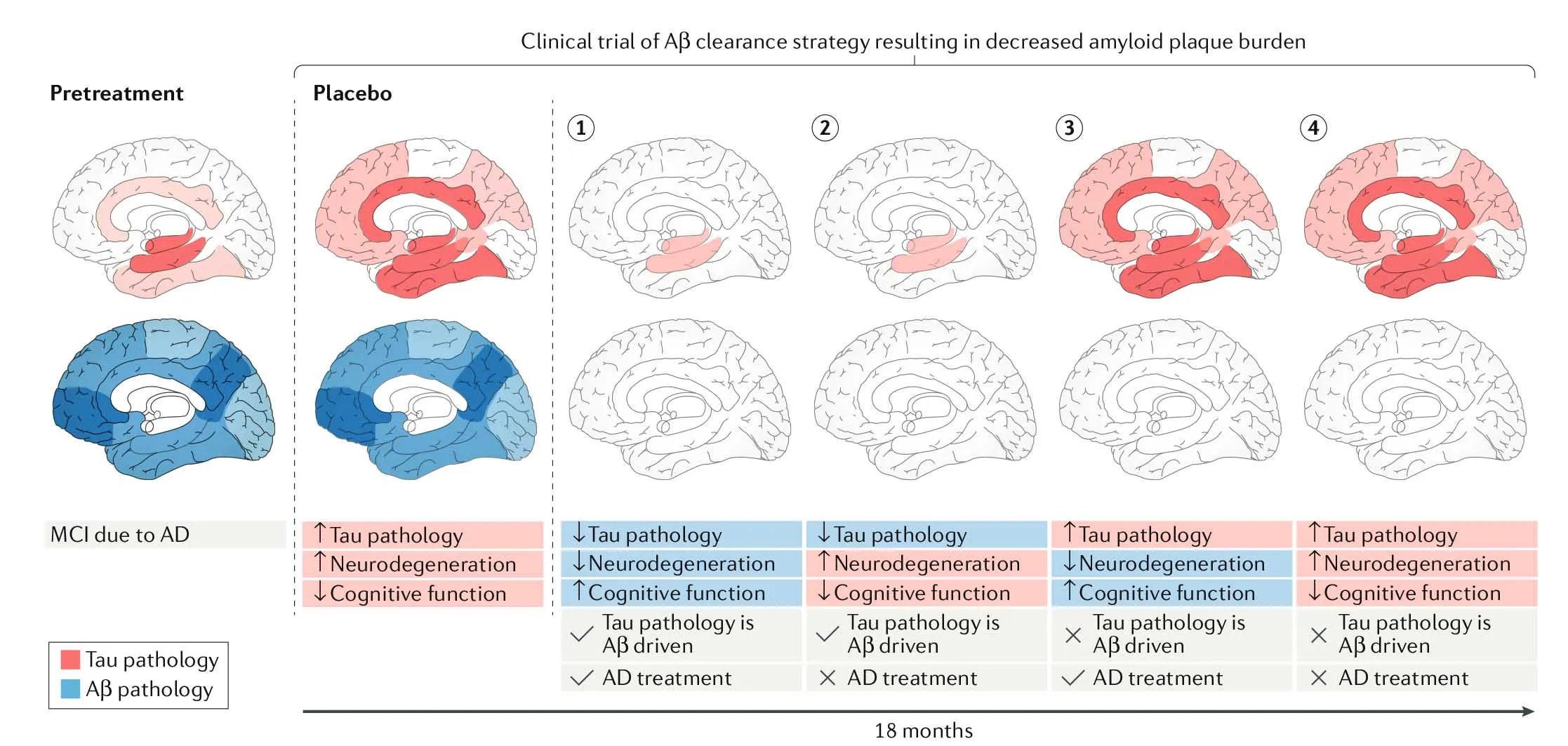
Conclusion
Almost 30 years after the formulation of the amyloid cascade hypothesis, the research framework on Alzheimer’s disease is impressively multifaceted. The focus on amyloid is now beginning to gradually shift towards new promising approaches directed against other elements of the disease, such as CNS inflammation, brain insulin resistance and tau aggregation (Panza et al., 2019).
Pursuing alternative hypotheses is a key motor in scientific discovery, and in a field as serious and complex as this, with countless open questions remaining, it is almost an imperative.
References
Benilova, I., Karran, E., & De Strooper, B. (2012). The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nature Neuroscience, 15(3), 349–357. https://doi.org/10.1038/nn.3028
Bloom, G. S. (2014). Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurology, 71(4), 505–508. https://doi.org/10.1001/jamaneurol.2013.5847
Brothers, H. M., Gosztyla, M. L., & Robinson, S. R. (2018). The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Frontiers in Aging Neuroscience, 10. https://doi.org/10.3389/fnagi.2018.00118
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., … Semagacestat Study Group. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. The New England Journal of Medicine, 369(4), 341–350. https://doi.org/10.1056/NEJMoa1210951
Hardy, J., & Higgins, G. (1992). Alzheimer’s disease: The amyloid cascade hypothesis. Science, 256(5054), 184–185. https://doi.org/10.1126/science.1566067
Hardy, John. (2009). The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal: Amyloid hypothesis for Alzheimer’s disease. Journal of Neurochemistry, 110(4), 1129–1134. https://doi.org/10.1111/j.1471-4159.2009.06181.x
Herrup, K. (2015). The case for rejecting the amyloid cascade hypothesis. Nature Neuroscience, 18(6), 794–799. https://doi.org/10.1038/nn.4017
Jack, C. R., Wiste, H. J., Schwarz, C. G., Lowe, V. J., Senjem, M. L., Vemuri, P., … Petersen, R. C. (2018). Longitudinal tau PET in ageing and Alzheimer’s disease. Brain, 141(5), 1517–1528. https://doi.org/10.1093/brain/awy059
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., … Stefansson, K. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature, 488(7409), 96–99. https://doi.org/10.1038/nature11283
Ju, Y.-E. S., Ooms, S. J., Sutphen, C., Macauley, S. L., Zangrilli, M. A., Jerome, G., … Holtzman, D. M. (2017). Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain, 140(8), 2104–2111. https://doi.org/10.1093/brain/awx148
Kametani, F., & Hasegawa, M. (2018). Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Frontiers in Neuroscience, 12, 25. https://doi.org/10.3389/fnins.2018.00025
Kawas, C., Gray, S., Brookmeyer, R., Fozard, J., & Zonderman, A. (2000). Age-specific incidence rates of Alzheimer’s disease: The Baltimore longitudinal study of aging. Neurology, 54(11), 2072–2077. https://doi.org/10.1212/WNL.54.11.2072
Kovacs, G. G. (2018). Chapter 25—Tauopathies. In G. G. Kovacs & I. Alafuzoff (A c. Di), Handbook of Clinical Neurology (page. 355–368). https://doi.org/10.1016/B978-0-12-802395-2.00025-0
Kumar-Singh, S., Theuns, J., Van Broeck, B., Pirici, D., Vennekens, K., Corsmit, E., … Van Broeckhoven, C. (2006). Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Human Mutation, 27(7), 686–695. https://doi.org/10.1002/humu.20336
Liu, C.-C., Kanekiyo, T., Xu, H., & Bu, G. (2013). Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nature Reviews Neurology, 9(2), 106–118. https://doi.org/10.1038/nrneurol.2012.263
Marklund, N., Farrokhnia, N., Hånell, A., Vanmechelen, E., Enblad, P., Zetterberg, H., … Hillered, L. (2014). Monitoring of β-amyloid dynamics after human traumatic brain injury. Journal of Neurotrauma, 31(1), 42–55. https://doi.org/10.1089/neu.2013.2964
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., & Cummings, J. L. (2015). Alzheimer’s disease. Nature Reviews Disease Primers, 1(1), 1–18. https://doi.org/10.1038/nrdp.2015.56
Ossenkoppele, R., Schonhaut, D. R., Schöll, M., Lockhart, S. N., Ayakta, N., Baker, S. L., … Rabinovici, G. D. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain, 139(5), 1551–1567. https://doi.org/10.1093/brain/aww027
Panza, F., Lozupone, M., Logroscino, G., & Imbimbo, B. P. (2019). A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nature Reviews. Neurology, 15(2), 73–88. https://doi.org/10.1038/s41582-018-0116-6
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M., West, R. J., Barber, P. C., & Butler, A. C. (1998). Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Annals of Neurology, 43(3), 380–383. https://doi.org/10.1002/ana.410430316
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerrière, A., Vital, A., … Campion, D. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature Genetics, 38(1), 24–26. https://doi.org/10.1038/ng1718
Sahyouni, R., Verma, A., & Chen, J. (2016). Alzheimer’s disease decoded: The history, present, and future of Alzheimer’s disease and dementia. https://doi.org/10.1142/10023
Selkoe, D. J., & Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine, 8(6), 595–608. https://doi.org/10.15252/emmm.201606210
van der Kant, R., Goldstein, L. S. B., & Ossenkoppele, R. (2019). Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nature Reviews Neuroscience, 1–15. https://doi.org/10.1038/s41583-019-0240-3
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., … Masters, C. L. (2013). Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. The Lancet Neurology, 12(4), 357–367. https://doi.org/10.1016/S1474-4422(13)70044-9
Waldemar, G., & Burns, A. (A c. Di). (2017). Alzheimer’s Disease (Second Edition). Oxford, New York: Oxford University Press.
Wisniewski, K. E., Wisniewski, H. M., & Wen, G. Y. (1985). Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Annals of Neurology, 17(3), 278–282. https://doi.org/10.1002/ana.410170310