Introduction
With a lifetime prevalence estimated between 0.3 and 0.7 % (van Os & Kapur, 2009), Schizophrenia is one of the most severe and disabling psychiatric disorders. Usually diagnosed between 15-35 years of age, it has a profound impact both on society and on the individual: sufferers show lifelong impairment in social functioning resistant to remission from psychotic symptoms (Thornicroft et al., 2004) and up to 50 % higher than average mortality rates, caused both by unhealthy lifestyles and complications correlated with the disorder or its treatment (Pillinger, D’Ambrosio, McCutcheon & Howes, 2019).
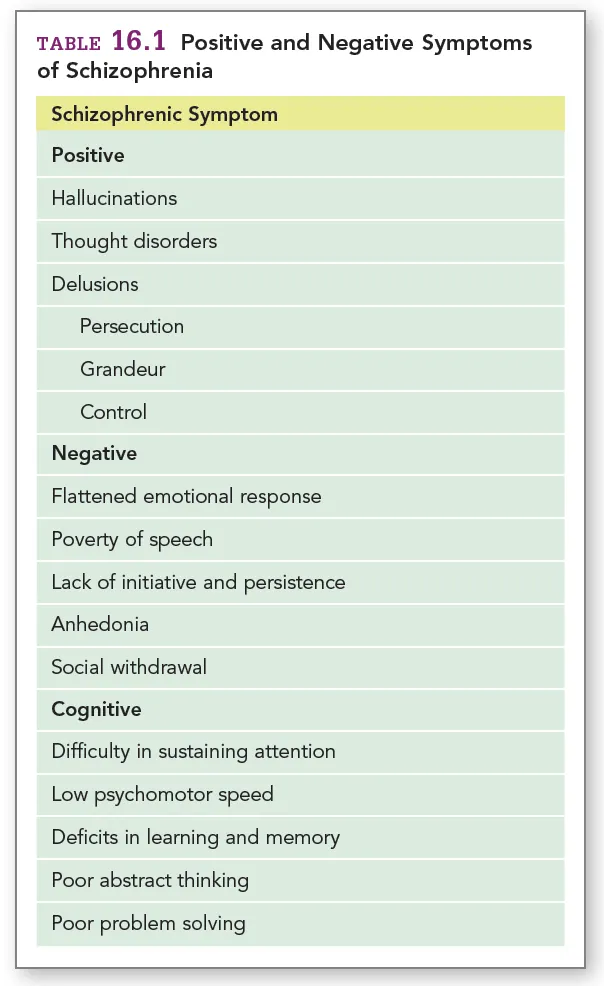
Schizophrenia presents itself as a complex syndrome with a heterogenous combination of symptoms ranging along a gradient of psychopathology, as reported in the Diagnostic and Statistical Manual of Mental Disorders (5th ed.; DSM-5; American Psychiatric Association, pp. 87-123), which reserves a chapter to “Schizophrenia spectrum disorders”. The symptoms are commonly classified in three categories: positive, negative and cognitive (Mueser & McGurk, 2004; summarised in Table 1), with cognitive symptoms now recognised as the prominent feature of the disease (Kahn & Keefe, 2013), resisting antipsychotic treatment and better predicting social functioning (Uno & Coyle, 2019). With a strong heritability, estimated around 79% (Hilker et al., 2018), the disorder is thought to emerge from the interplay between genetic, developmental and environmental factors.
Gross brain pathology is not a core feature of Schizophrenia (Kahn et al., 2015): a more subtle kind of pathology can be viewed under a neural circuit perspective, following the now widely accepted “dysconnectivity hypothesis” (Friston, 1998). The schizophrenic brain is in fact characterised by abnormal structural and functional connectivity across distributed neural networks both at the macroscopic and the microscopic level, resulting in significant changes in the way information is processed and integrated between and within different regions (Lynall et al., 2010; van den Heuvel & Fornito, 2014).
Different brain areas and neurotransmitter pathways play a role in this dysfunction: in this essay, I am going to explore some of the main actors, to better understand the direction of treatment efforts.
The basal ganglia
The basal ganglia are a set of functionally related nuclei in the brain implicated in different cortical-subcortical circuits modulating motor, cognitive and emotional behaviour (Perez-Costas, Melandez-Ferro & Roberts, 2010). A comprehensive description of the anatomy and connectivity patterns of the basal ganglia is well beyond the scope of this essay. A simplified version of the classical model will be considered, and it is illustrated in figure 1.
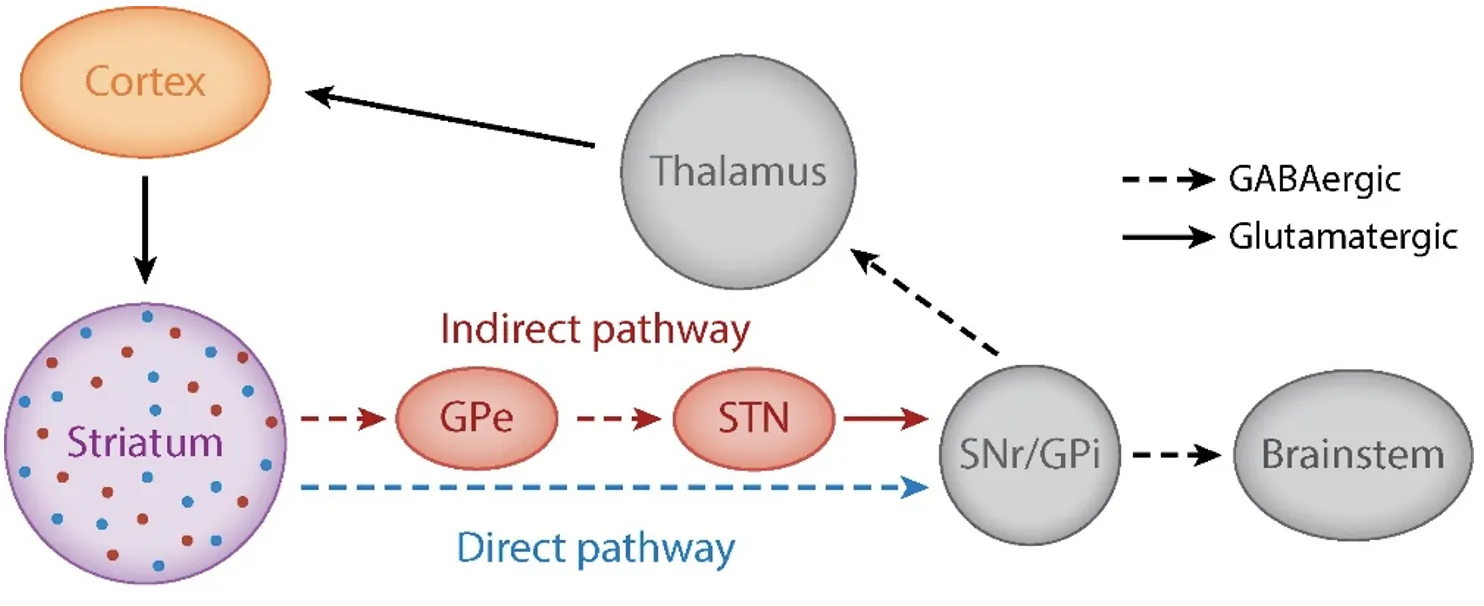
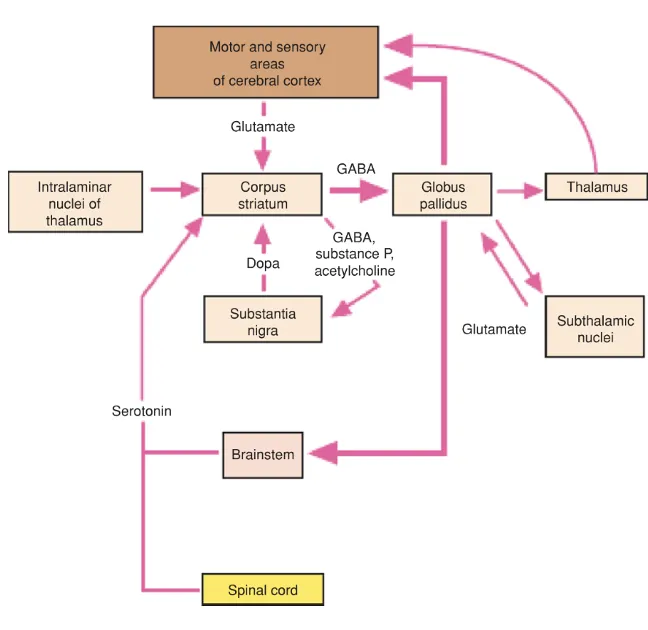
Notably, dysfunctional interactions between the cortex and the striatum are often implicated in psychiatric conditions (Gunaydin & Kreitzer, 2016); indeed, as we will see, the dorsal striatum and the dorsolateral prefrontal cortex (dlPFC) are critical sites of dysfunction in Schizophrenia. What is also crucial to our discussion is the complex interaction between different neurotransmitter systems between and within the basal ganglia, the prefrontal cortex and the brain stem, which are schematised in figure 3. Alterations in this web of interactions are at the root of schizophrenia pathology.
Let us look more in-depth at the roles of different transmitters.
Dopamine
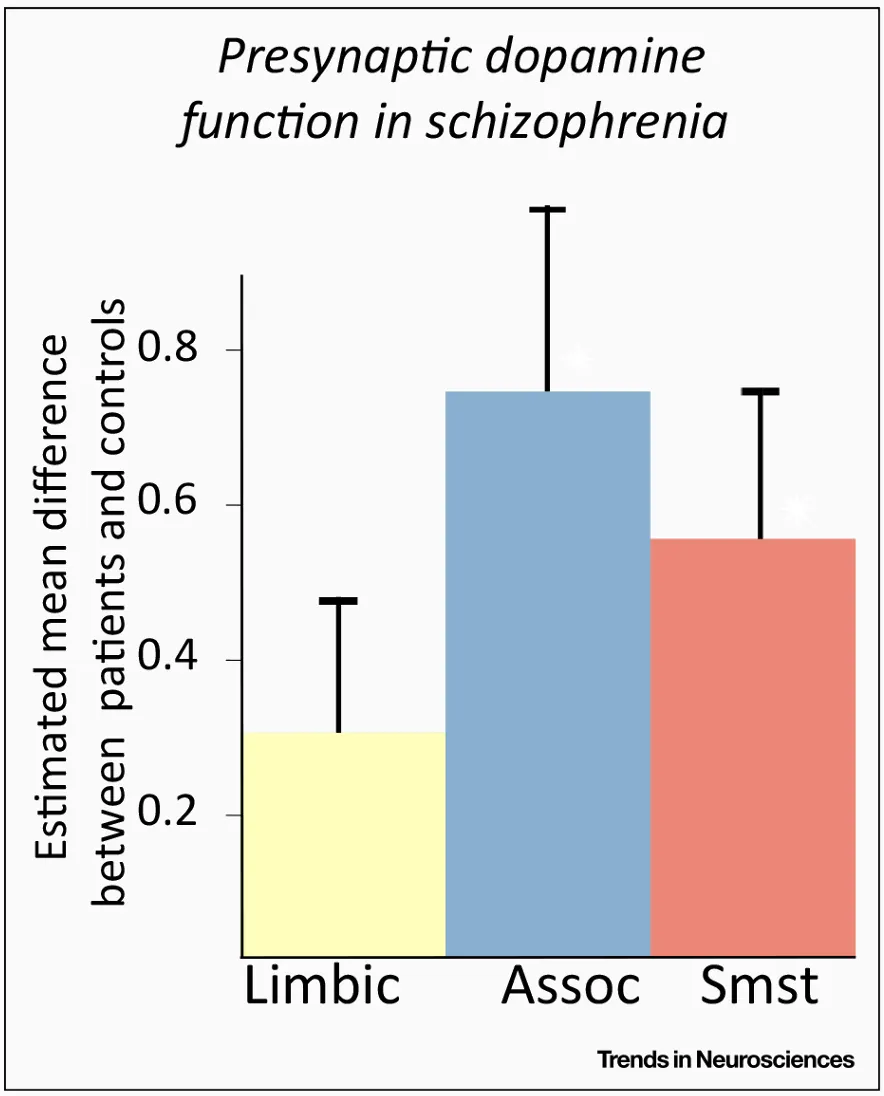
The dopaminergic system in the basal ganglia has a major role in arousal, attribution of saliency to stimuli, motivation and reinforcement learning through predictive coding of reward stimuli (Kapur, 2003; Schultz, Dayan & Montague, 1997; Perez-Costas, Melandez-Ferro & Roberts, 2010). The finding that dopamine synthesis and release in the striatum is increased in psychotic patients is very robust, especially when looking at the dorsal – or associative – striatum (McCutcheon, Beck, Jauhar & Howes, 2018), which has recently been recognised as the primary site of dopamine dysfunction, contrary to long-held theories pointing to the mesolimbic striatum (McCutcheon, Abi-Dargham & Howes, 2019; figure 3).
The dopamine hypothesis of Schizophrenia, according to which central to the pathogenesis of the disease is a dysregulation of the brain’s dopamine system, especially in the mesolimbic striatum, is one of the most enduring and influential theories in psychiatry.
The hypothesis was initially born from the observation that neuroleptic drugs interfere with dopamine function in the basal ganglia and corroborated by results on studies on the mechanisms of actions of psychomotor stimulants such as amphetamine (Baumeister & Francis, 2002).
After many years from its formulation, however, many factors point towards the insufficiency of the dopamine hypothesis. Even if every licensed pharmacological treatment of schizophrenia targets in some way the dopamine system (McCutcheon, Abi-Dargham & Howes, 2019), and has good effects in reducing positive symptoms (Sommer et al., 2012), antipsychotic treatments have negligible effects on cognitive symptoms (Lim et al., 2016). This is a significant shortcoming when considering that cognitive symptoms are the best predictors of disability and a core feature of the disease, developing prior to the onset of psychosis (Reichenberg et al., 2010). Thus, a broader perspective is needed.
Glutamate
Evidence for the involvement of glutamate neurotransmission, and especially through N-methyl-D-aspartate (NMDA) glutamate receptors hypofunction in Schizophrenia pathophysiology come from various sources. First, there is the observation that the non-competitive NMDA antagonists ketamine and phencyclidine can induce psychotic episodes that are also accompanied by the negative and cognitive symptoms seen in Schizophrenia (Moghaddam & Krystal, 2012). This is opposed to dopaminergic agonists, which can induce psychotic states but do not reproduce a whole spectrum of trait phenomena (Adams, Stephan, Brown, Frith & Friston, 2013). Second, on the genetics side, genome-wide association studies (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) and large-scale copy-number variants studies (Pocklington et al., 2015) find consistent relationships between mutations in genes involved in glutamatergic transmission and increased risk of Schizophrenia. Third, post-mortem neurochemical studies also find altered expression of NMDA receptors (Catts, Lai, Weickert, Weickert & Catts, 2016) and receptor-associated postsynaptic density proteins (Kristiansen, Beneyto, Haroutunian & Meador-Woodruff, 2006) in subjects with Schizophrenia.
NMDA receptors have received special attention for their unique biophysical properties, which can make important contributions to the dynamics of neuronal networks and participate in essential mechanisms related to synaptic plasticity. Just to give two examples, NMDA receptor activity in the dlPFC is thought to be key to working memory functioning (Durstewitz, 2009), and NMDA receptor properties are thought to be central determinants of synaptic gain in neurons reporting prediction errors. The latter is a central mechanism in computational theories of perception and action, with the potential to explain psychotic episodes in terms of misattribution of salience to stimuli (Adams et al., 2013).
The promise of targeting glutamatergic dysfunctions would be of finally treating cognitive symptoms efficiently. Unfortunately, treatment based on this glutamate hypothesis of Schizophrenia has not yet been as successful as expected. NMDA receptors co-agonists and metabotropic glutamate receptors agonists are both being studied extensively. However, early results are mixed, suggesting that perhaps a more precise sub-grouping of patients with more pronounced glutamatergic pathology could guarantee better treatment outcomes and fewer inconsistencies across studies (Uno & Coyle, 2019).
GABAergic interneurons
Converging evidence now supports the idea that NMDA receptors hypofunction exerts its most significant effects on GABAergic interneurons (Nakazawa et al., 2011). Evidence of reduced GABAergic neuronal activity is a consistent finding in patients with Schizophrenia, even in post-mortem studies (Schmidt & Mirnics, 2015). The dysfunction is particularly relevant for fast-spiking parvalbumin(PV)-containing interneurons, which have a critical role in the dlPFC in the generation of gamma-frequency oscillations related to working memory activity, and more broadly in the synchronisation of local populations of cortical neurons (Lewis & González-Burgos, 2008). Interestingly, PV-containing GABAergic interneurons also have a protracted development period, dependent on NMDA stimulation and culminating in adolescence (Belforte et al., 2010), a feature consistent with the progressive emergence of Schizophrenia during the maturation of neural networks (Nakazawa et al., 2011). How GABAergic interneurons fit in the pathophysiology of the disease is summarised in Figure 4.
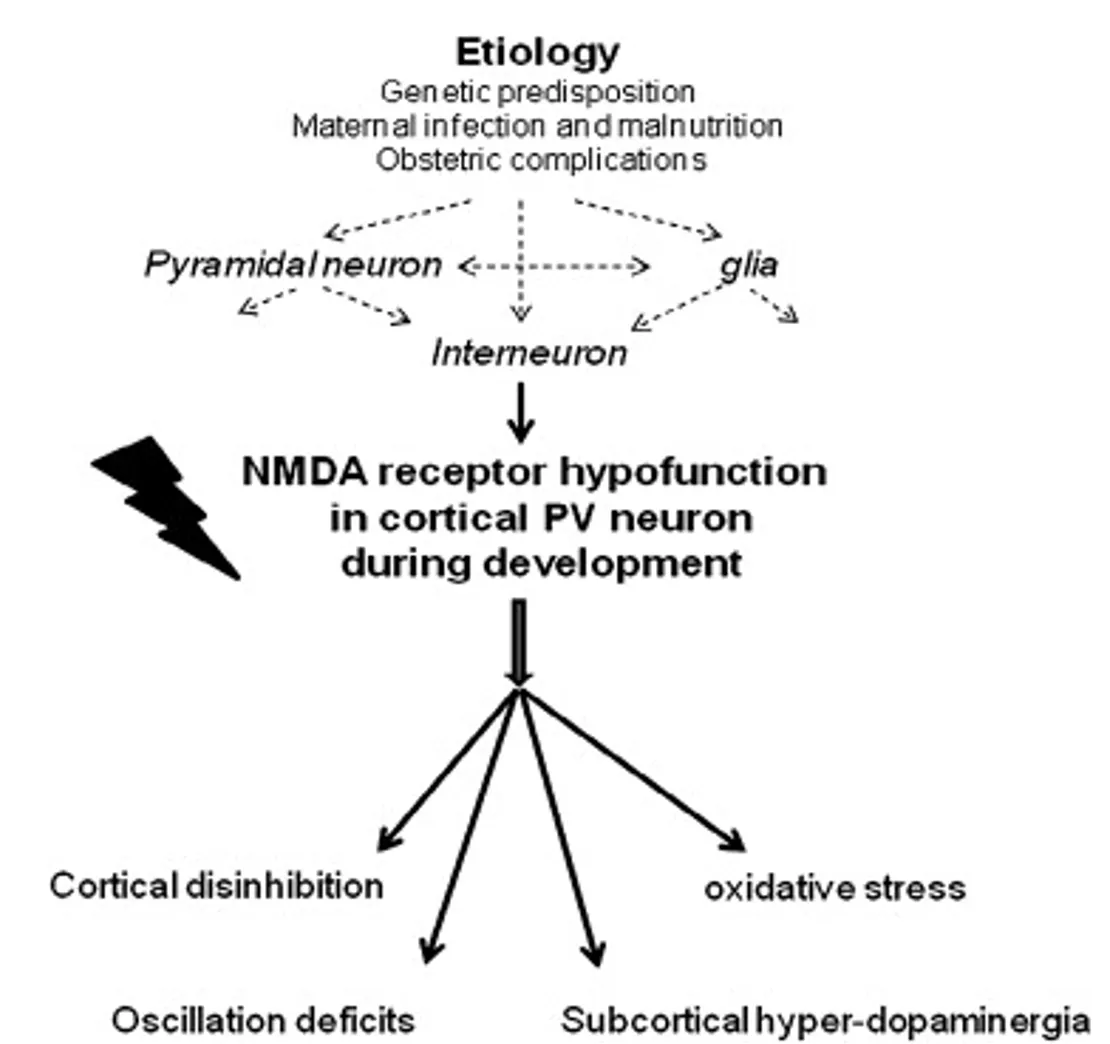
As far as treatment is concerned, this GABAergic dysfunction is difficult to target directly. To begin with, there is the interesting observation that GABA agonists have been occasionally reported to induce psychosis, while antagonists are by no means antipsychotic (Kahn et al., 2015). However, GABA agonists such as benzodiazepines have been used clinically alongside antipsychotics to ameliorate Schizophrenia’s positive symptoms (Wassef, Baker & Kochan, 2003). Selective drugs that target GABA receptor subtypes directly to treat cognitive symptoms of Schizophrenia are being evaluated (Gill & Grace, 2014), but none are in clinical trials.
Acetylcholine
The clinical observation that a lot of schizophrenia patients smoke heavily and do so to ameliorate negative symptoms and reduce antipsychotics’ side effects (Sagud et al., 2009), has raised some interesting questions on the possible roles of acetylcholine in the pathophysiology and treatment of the disease. Post-mortem and neuroimaging studies do indeed suggest cholinergic dysfunctions both in the cortex and in the striatum (Gibbons & Dean, 2016) of patients with Schizophrenia. Moreover, cholinergic interneurons are recently emerging as critical players in striatal function, especially in cortical control of dopamine transmission (Kosillo, Zhang, Threlfell & Cragg, 2016). Agonists of the nicotinic and muscarinic acetylcholine receptors are thus under exploration, being promising treatment mechanisms for cognitive and negative symptoms, supposedly even able to modulate dopamine release directly in the dorsal striatum, where it would be most in need of regularisation (McCutcheon, Abi-Dargham & Howes, 2019).
Notable mentions
Other notable mentions for their role in the research on Schizophrenia are serotonin and inflammation.
Serotonin hypotheses of Schizophrenia were among the first to be formulated (Aghajanian & Marek, 2000). However, the observation that second-generation antipsychotics, by including serotonergic antagonists, reduce extrapyramidal side-effects but do not provide better efficacy against symptoms, has seen a decline in hypothesis regarding serotonin directly in the pathogenesis of the disease. Nonetheless, the role of serotonin is still being explored, even as a potential therapeutic target (Abi-Dargham, 2007) for the negative and cognitive symptoms.
The role of inflammation, on the other hand, has not been explored until recently (Feigenson, Kusnecov & Silverstein, 2014), but it is receiving a lot of attention, especially for treatment in the early phases of the disease (Yang & Tsai, 2017).
Overall evaluation
Table 2 summarises the new directions for schizophrenia treatment emerging from our discussion. To put everything in context, figure 5 depicts the image of the schizophrenic brain we have been portraying up to this point.
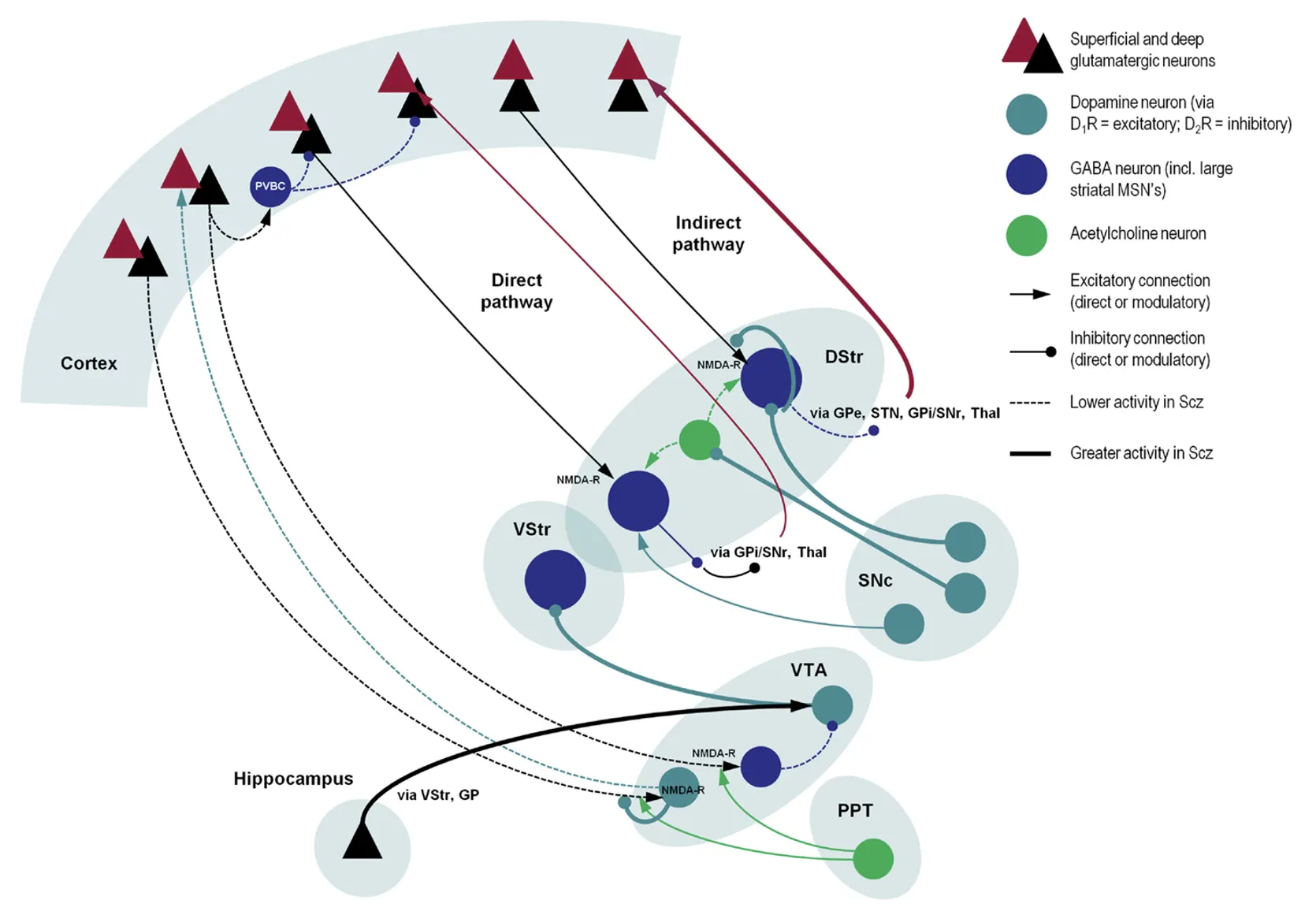
Clearly, the picture of the pathophysiology we sketched is tentative and incomplete. Research in this area is very complicated, primarily because it is plagued by many confounds and difficulties (Perez-Costas, Melandez-ferro & Roberts, 2010): schizophrenia symptoms in itself range across a heterogeneous spectrum, making diagnosis and categorisation of patients a critical process; medication and substance abuse history can impact treatment outcomes, and patients have a very diverse response to treatment anyway; critically, no complete animal models exist for the disease, even if continuous effort is being made in this direction (Kesby, Eyles, McGrath & Scott, 2018).
Conclusion
Antipsychotics targeting the dopamine system had a massive impact on the clinical treatment of patients with Schizophrenia from the 1950s onwards, by significantly reducing the need for hospitalisation and eliminating other questionable forms of intervention (such as electroconvulsive therapy and prefrontal lobotomy). However, these drugs are far from perfect, in that they suppress rather than eradicate the symptoms, cause numerous side effects and do not treat cognitive and negative symptoms very efficiently (Davey, 2014; pp. 270-272). Clearly, a move beyond the dopamine hypothesis of Schizophrenia is needed by looking through a circuits perspective at the complex and multi-faceted interactions between different neurotransmitter systems. This will only be possible through the simultaneous advancement of many interconnected fields in neuroscience, from detailed anatomy of microcircuit structures to functional imaging of broader networks. There is still a long way to go, but the field is very active, and the results so far undoubtedly promising.
References
Abi-Dargham, A. (2007). Alterations of serotonin transmission in schizophrenia. International Review of Neurobiology, 78, 133–164. https://doi.org/10.1016/S0074-7742(06)78005-9
Adams, R. A., Stephan, K. E., Brown, H. R., Frith, C. D., & Friston, K. J. (2013). The computational anatomy of psychosis. Frontiers in Psychiatry, 4. https://doi.org/10.3389/fpsyt.2013.00047
Aghajanian, G. K., & Marek, G. J. (2000). Serotonin model of schizophrenia: Emerging role of glutamate mechanisms. Brain Research. Brain Research Reviews, 31(2–3), 302–312. https://doi.org/10.1016/s0165-0173(99)00046-6
American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (Fifth Edition). American Psychiatric Association. https://doi.org/10.1176/appi.books.9780890425596
Baumeister, A. A., & Francis, J. L. (2002). Historical development of the dopamine hypothesis of schizophrenia. Journal of the History of the Neurosciences, 11(3), 265–277. https://doi.org/10.1076/jhin.11.3.265.10391
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z., Yu, G., Li, Y., Quinlan, E. M., & Nakazawa, K. (2010a). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nature Neuroscience, 13(1), 76–83. https://doi.org/10.1038/nn.2447
Belforte, J. E., Zsiros, V., Sklar, E. R., Jiang, Z., Yu, G., Li, Y., Quinlan, E. M., & Nakazawa, K. (2010b). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nature Neuroscience, 13(1), 76–83. https://doi.org/10.1038/nn.2447
Carlson, N. R. (2013). Physiology of behavior (Eleventh ed). Pearson.
Catts, V. S., Lai, Y. L., Weickert, C. S., Weickert, T. W., & Catts, S. V. (2016). A quantitative review of the postmortem evidence for decreased cortical N-methyl-D-aspartate receptor expression levels in schizophrenia: How can we link molecular abnormalities to mismatch negativity deficits? Biological Psychology, 116, 57–67. https://doi.org/10.1016/j.biopsycho.2015.10.013
Davey, G. (2014). Psychopathology: Research, assessment and treatment in clinical psychology (Second edition). The British Psychological Society ; Wiley.
Durstewitz, D. (2009). Implications of synaptic biophysics for recurrent network dynamics and active memory. Neural Networks: The Official Journal of the International Neural Network Society, 22(8), 1189–1200. https://doi.org/10.1016/j.neunet.2009.07.016
Feigenson, K. A., Kusnecov, A. W., & Silverstein, S. M. (2014). Inflammation and the two-hit hypothesis of schizophrenia. Neuroscience and Biobehavioral Reviews, 38, 72–93. https://doi.org/10.1016/j.neubiorev.2013.11.006
Friston, K. J. (1998). The disconnection hypothesis. Schizophrenia Research, 30(2), 115–125. https://doi.org/10.1016/s0920-9964(97)00140-0
Gibbons, A., & Dean, B. (2016). The cholinergic system: An emerging drug target for schizophrenia. Current Pharmaceutical Design, 22(14), 2124–2133. https://doi.org/10.2174/1381612822666160127114010
Gill, K. M., & Grace, A. A. (2014). The role of α5 GABAA receptor agonists in the treatment of cognitive deficits in schizophrenia. Current pharmaceutical design, 20(31), 5069–5076.
Gunaydin, L. A., & Kreitzer, A. C. (2016). Cortico–basal ganglia circuit function in psychiatric disease. Annual Review of Physiology, 78(1), 327–350. https://doi.org/10.1146/annurev-physiol-021115-105355
Hilker, R., Helenius, D., Fagerlund, B., Skytthe, A., Christensen, K., Werge, T. M., Nordentoft, M., & Glenthøj, B. (2018). Heritability of schizophrenia and schizophrenia spectrum based on the nationwide danish twin register. Biological Psychiatry, 83(6), 492–498. https://doi.org/10.1016/j.biopsych.2017.08.017
Kahn, R. S., & Keefe, R. S. E. (2013). Schizophrenia is a cognitive illness: Time for a change in focus. JAMA Psychiatry, 70(10), 1107–1112. https://doi.org/10.1001/jamapsychiatry.2013.155
Kahn, R. S., Sommer, I. E., Murray, R. M., Meyer-Lindenberg, A., Weinberger, D. R., Cannon, T. D., O’Donovan, M., Correll, C. U., Kane, J. M., van Os, J., & Insel, T. R. (2015). Schizophrenia. Nature Reviews Disease Primers, 1(1), 1–23. https://doi.org/10.1038/nrdp.2015.67
Kapur, S. (2003). Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. The American Journal of Psychiatry, 160(1), 13–23. https://doi.org/10.1176/appi.ajp.160.1.13
Kesby, J. P., Eyles, D. W., McGrath, J. J., & Scott, J. G. (2018). Dopamine, psychosis and schizophrenia: The widening gap between basic and clinical neuroscience. Translational Psychiatry, 8(1), 1–12. https://doi.org/10.1038/s41398-017-0071-9
Kosillo, P., Zhang, Y.-F., Threlfell, S., & Cragg, S. J. (2016). Cortical control of striatal dopamine transmission via striatal cholinergic interneurons. Cerebral Cortex (New York, N.Y.: 1991), 26(11), 4160–4169. https://doi.org/10.1093/cercor/bhw252
Kristiansen, L. V., Beneyto, M., Haroutunian, V., & Meador-Woodruff, J. H. (2006). Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Molecular Psychiatry, 11(8), 737–747, 705. https://doi.org/10.1038/sj.mp.4001844
Lewis, D. A., & González-Burgos, G. (2008). Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 33(1), 141–165. https://doi.org/10.1038/sj.npp.1301563
Lim, J., Lee, S.-A., Lam, M., Rapisarda, A., Kraus, M., Keefe, R. S. E., & Lee, J. (2016). The relationship between negative symptom subdomains and cognition. Psychological Medicine, 46(10), 2169–2177. https://doi.org/10.1017/S0033291716000726
Lynall, M.-E., Bassett, D. S., Kerwin, R., McKenna, P. J., Kitzbichler, M., Muller, U., & Bullmore, E. (2010). Functional connectivity and brain networks in schizophrenia. Journal of Neuroscience, 30(28), 9477–9487. https://doi.org/10.1523/JNEUROSCI.0333-10.2010
McCutcheon, R. A., Abi-Dargham, A., & Howes, O. D. (2019a). Schizophrenia, dopamine and the striatum: From biology to symptoms. Trends in Neurosciences, 42(3), 205–220. https://doi.org/10.1016/j.tins.2018.12.004
McCutcheon, R. A., Abi-Dargham, A., & Howes, O. D. (2019b). Schizophrenia, dopamine and the striatum: From biology to symptoms. Trends in Neurosciences, 42(3), 205–220. https://doi.org/10.1016/j.tins.2018.12.004
McCutcheon, R., Beck, K., Jauhar, S., & Howes, O. D. (2018). Defining the locus of dopaminergic dysfunction in schizophrenia: A meta-analysis and test of the mesolimbic hypothesis. Schizophrenia Bulletin, 44(6), 1301–1311. https://doi.org/10.1093/schbul/sbx180
Moghaddam, B., & Krystal, J. H. (2012). Capturing the angel in “angel dust”: Twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophrenia Bulletin, 38(5), 942–949. https://doi.org/10.1093/schbul/sbs075
Mueser, K. T., & McGurk, S. R. (2004). Schizophrenia. Lancet (London, England), 363(9426), 2063–2072. https://doi.org/10.1016/S0140-6736(04)16458-1
Nakazawa, K., Zsiros, V., Jiang, Z., Nakao, K., Kolata, S., Zhang, S., & Belforte, J. E. (2012). GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology, 62(3), 1574–1583. https://doi.org/10.1016/j.neuropharm.2011.01.022
Perez-Costas, E., Melendez-Ferro, M., & Roberts, R. C. (2010). Basal ganglia pathology in schizophrenia: Dopamine connections and anomalies. Journal of Neurochemistry, 113(2), 287–302. https://doi.org/10.1111/j.1471-4159.2010.06604.x
Pillinger, T., D’Ambrosio, E., McCutcheon, R., & Howes, O. D. (2019). Is psychosis a multisystem disorder? A meta-review of central nervous system, immune, cardiometabolic, and endocrine alterations in first-episode psychosis and perspective on potential models. Molecular Psychiatry, 24(6), 776–794. https://doi.org/10.1038/s41380-018-0058-9
Pocklington, A. J., Rees, E., Walters, J. T. R., Han, J., Kavanagh, D. H., Chambert, K. D., Holmans, P., Moran, J. L., McCarroll, S. A., Kirov, G., O’Donovan, M. C., & Owen, M. J. (2015). Novel findings from cnvs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron, 86(5), 1203–1214. https://doi.org/10.1016/j.neuron.2015.04.022
Reichenberg, A., Caspi, A., Harrington, H., Houts, R., Keefe, R. S. E., Murray, R. M., Poulton, R., & Moffitt, T. E. (2010). Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: A 30-year study. The American Journal of Psychiatry, 167(2), 160–169. https://doi.org/10.1176/appi.ajp.2009.09040574
Sagud, M., Mihaljević-Peles, A., Mück-Seler, D., Pivac, N., Vuksan-Cusa, B., Brataljenović, T., & Jakovljević, M. (2009). Smoking and schizophrenia. Psychiatria Danubina, 21(3), 371–375.
Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511(7510), 421–427. https://doi.org/10.1038/nature13595
Schmidt, M. J., & Mirnics, K. (2015). Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 40(1), 190–206. https://doi.org/10.1038/npp.2014.95
Schultz, W., Dayan, P., & Montague, P. R. (1997). A neural substrate of prediction and reward. Science (New York, N.Y.), 275(5306), 1593–1599. https://doi.org/10.1126/science.275.5306.1593
Sommer, I. E. C., Slotema, C. W., Daskalakis, Z. J., Derks, E. M., Blom, J. D., & van der Gaag, M. (2012). The treatment of hallucinations in schizophrenia spectrum disorders. Schizophrenia Bulletin, 38(4), 704–714. https://doi.org/10.1093/schbul/sbs034
Splittgerber, R., & Snell, R. S. (2019). Snell’s clinical neuroanatomy (Eighth edition). Wolters Kluwer.
Thornicroft, G., Tansella, M., Becker, T., Knapp, M., Leese, M., Schene, A., Vazquez-Barquero, J. L., & EPSILON Study Group. (2004). The personal impact of schizophrenia in Europe. Schizophrenia Research, 69(2–3), 125–132. https://doi.org/10.1016/s0920-9964(03)00191-9
Uno, Y., & Coyle, J. T. (2019). Glutamate hypothesis in schizophrenia. Psychiatry and Clinical Neurosciences, 73(5), 204–215. https://doi.org/10.1111/pcn.12823
van den Heuvel, M. P., & Fornito, A. (2014). Brain networks in schizophrenia. Neuropsychology Review, 24(1), 32–48. https://doi.org/10.1007/s11065-014-9248-7
van Os, J., & Kapur, S. (2009). Schizophrenia. Lancet (London, England), 374(9690), 635–645. https://doi.org/10.1016/S0140-6736(09)60995-8
Wassef, A., Baker, J., & Kochan, L. D. (2003). GABA and schizophrenia: A review of basic science and clinical studies. Journal of Clinical Psychopharmacology, 23(6), 601–640. https://doi.org/10.1097/01.jcp.0000095349.32154.a5
Yang, A., & Tsai, S.-J. (2017). New targets for schizophrenia treatment beyond the dopamine hypothesis. International Journal of Molecular Sciences, 18(8), 1689. https://doi.org/10.3390/ijms18081689